1
2
3
4
5
6
7
8
9
10
11
12
13
14
15
16
17
18
19
20
21
22
23
24
25
26
27
28
29
30
31
32
33
34
35
36
37
38
39
40
41
42
43
44
45
46
47
48
49
50
51
52
53
54
55
56
57
58
59
60
61
62
63
64
65
66
67
68
69
70
71
72
73
74
75
76
77
78
79
80
81
82
83
84
85
86
87
88
89
90
91
92
93
94
95
96
97
98
99
100
101
102
103
104
105
106
107
108
109
110
111
112
113
114
115
116
117
118
119
120
121
122
123
124
125
126
127
128
129
130
131
132
133
134
135
136
137
138
139
140
141
142
143
144
145
146
147
148
149
150
151
152
153
154
155
156
157
158
159
160
161
162
163
164
165
166
167
168
169
170
171
172
173
174
175
176
177
178
179
180
181
182
183
184
185
186
187
188
189
190
191
192
193
194
195
196
197
198
199
200
201
202
203
204
205
206
207
208
209
210
211
212
213
214
215
216
217
218
219
220
221
222
223
224
225
226
227
228
229
230
231
232
233
234
235
236
237
238
239
240
241
//! rebop is a fast stochastic simulator for well-mixed chemical
//! reaction networks.
//!
//! Performance and ergonomics are taken very seriously. For this reason,
//! two independent APIs are provided to describe and simulate reaction
//! networks:
//!
//! * a macro-based DSL implemented by [`define_system`], usually the
//! most efficient, but that requires to compile a rust program;
//! * a function-based API implemented by the module [`gillespie`], also
//! available through Python bindings. This one does not require a rust
//! compilation and allows the system to be defined at run time. It is
//! typically 2 or 3 times slower than the macro DSL, but still faster
//! than all other software tried.
//!
//! # The macro DSL
//!
//! It currently only supports reaction rates defined by the law of mass
//! action. The following macro defines a dimerization reaction network
//! naturally:
//!
//! ```rust
//! use rebop::define_system;
//! define_system! {
//! r_tx r_tl r_dim r_decay_mRNA r_decay_prot;
//! Dimers { gene, mRNA, protein, dimer }
//! transcription : gene => gene + mRNA @ r_tx
//! translation : mRNA => mRNA + protein @ r_tl
//! dimerization : 2 protein => dimer @ r_dim
//! decay_mRNA : mRNA => @ r_decay_mRNA
//! decay_protein : protein => @ r_decay_prot
//! }
//! ```
//!
//! To simulate the system, put this definition in a rust code file and
//! instantiate the problem, set the parameters, the initial values, and
//! launch the simulation:
//!
//! ```rust
//! # use rebop::define_system;
//! # define_system! {
//! # r_tx r_tl r_dim r_decay_mRNA r_decay_prot;
//! # Dimers { gene, mRNA, protein, dimer }
//! # transcription : gene => gene + mRNA @ r_tx
//! # translation : mRNA => mRNA + protein @ r_tl
//! # dimerization : 2 protein => dimer @ r_dim
//! # decay_mRNA : mRNA => @ r_decay_mRNA
//! # decay_protein : protein => @ r_decay_prot
//! # }
//! let mut problem = Dimers::new();
//! problem.r_tx = 25.0;
//! problem.r_tl = 1000.0;
//! problem.r_dim = 0.001;
//! problem.r_decay_mRNA = 0.1;
//! problem.r_decay_prot = 1.0;
//! problem.gene = 1;
//! problem.advance_until(1.0);
//! println!("t = {}: dimer = {}", problem.t, problem.dimer);
//! ```
//!
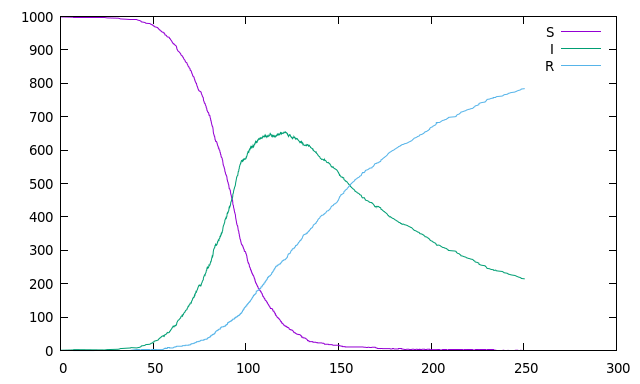
//! Or for the classic SIR example:
//!
//! ```rust
//! use rebop::define_system;
//!
//! define_system! {
//! r_inf r_heal;
//! SIR { S, I, R }
//! infection : S + I => 2 I @ r_inf
//! healing : I => R @ r_heal
//! }
//!
//! fn main() {
//! let mut problem = SIR::new();
//! problem.r_inf = 1e-4;
//! problem.r_heal = 0.01;
//! problem.S = 999;
//! problem.I = 1;
//! println!("time,S,I,R");
//! for t in 0..250 {
//! problem.advance_until(t as f64);
//! println!("{},{},{},{}", problem.t, problem.S, problem.I, problem.R);
//! }
//! }
//! ```
//!
//! which can produce an output similar to this one:
//!
//! 
//!
//! # Python bindings
//!
//! This API shines through the Python bindings which allow one to
//! define a model easily:
//!
//! ```python
//! import rebop
//!
//! sir = rebop.Gillespie()
//! sir.add_reaction(1e-4, ['S', 'I'], ['I', 'I'])
//! sir.add_reaction(0.01, ['I'], ['R'])
//! print(sir)
//!
//! ds = sir.run({'S': 999, 'I': 1}, tmax=250, nb_steps=250)
//! ```
//!
//! You can test this code by installing `rebop` from PyPI with
//! `pip install rebop`. To build the Python bindings from source,
//! the simplest is to clone this git repository and use `maturin
//! develop`.
//!
//! # The traditional API
//!
//! The function-based API underlying the Python package is also available
//! from Rust, if you want to be able to define models at run time (instead
//! of at compilation time with the macro DSL demonstrated above).
//! The SIR model is defined as:
//!
//! ```rust
//! use rebop::gillespie::{Gillespie, Rate};
//!
//! let mut sir = Gillespie::new([999, 1, 0], false);
//! // [ S, I, R]
//! // S + I => 2 I with rate 1e-4
//! sir.add_reaction(Rate::lma(1e-4, [1, 1, 0]), [-1, 1, 0]);
//! // I => R with rate 0.01
//! sir.add_reaction(Rate::lma(0.01, [0, 1, 0]), [0, -1, 1]);
//!
//! println!("time,S,I,R");
//! for t in 0..250 {
//! sir.advance_until(t as f64);
//! println!("{},{},{},{}", sir.get_time(), sir.get_species(0), sir.get_species(1), sir.get_species(2));
//! }
//! ```
//!
//! # Performance
//!
//! Performance is taken very seriously, and as a result, rebop
//! outperforms every other package and programming language that we
//! tried.
//!
//! *Disclaimer*: Most of this software currently contains much more
//! features than rebop (e.g. spatial models, custom reaction rates,
//! etc.). Some of these features might have required them to make
//! compromises on speed. Moreover, as much as we tried to keep the
//! comparison fair, some return too much or too little data, or write
//! them on disk. The baseline that we tried to approach for all these
//! programs is the following: *the model was just modified, we want
//! to simulate it `N` times and print regularly spaced measurement
//! points*. This means that we always include initialization or
//! (re-)compilation time if applicable. We think that it is the most
//! typical use-case of a researcher who works on the model. This
//! benchmark methods allows to record both the initialization time
//! (y-intercept) and the simulation time per simulation (slope).
//!
//! Many small benchmarks on toy examples are tracked to guide the
//! development. To compare the performance with other software,
//! we used a real-world model of low-medium size (9 species and 16
//! reactions): the Vilar oscillator (*Mechanisms of noise-resistance
//! in genetic oscillators*, Vilar et al., PNAS 2002). Here, we
//! simulate this model from `t=0` to `t=200`, reporting the state at
//! time intervals of `1` time unit.
//!
//! 
//!
//! We can see that rebop's macro DSL is the fastest of all, both in
//! time per simulation, and with compilation time included. The second
//! fastest is rebop's traditional API invoked by convenience through
//! the Python bindings.
//!
//! # Features to come
//!
//! * compartment volumes
//! * arbitrary reaction rates
//! * other SSA algorithms
//! * tau-leaping
//! * adaptive tau-leaping
//! * hybrid models (continuous and discrete)
//! * SBML
//! * CLI interface
//! * parameter estimation
//! * local sensitivity analysis
//! * parallelization
//!
//! # Features probably not to come
//!
//! * events
//! * space (reaction-diffusion systems)
//! * rule modelling
//!
//! # Benchmark ideas
//!
//! * DSMTS
//! * purely decoupled exponentials
//! * ring
//! * Toggle switch
//! * LacZ, LacY/LacZ (from STOCKS)
//! * Lotka Volterra, Michaelis--Menten, Network (from StochSim)
//! * G protein (from SimBiology)
//! * Brusselator / Oregonator (from Cellware)
//! * GAL, repressilator (from Dizzy)
//!
//! # Similar software
//!
//! ## Maintained
//!
//! * [GillesPy2](https://github.com/StochSS/GillesPy2)
//! * [STEPS](https://github.com/CNS-OIST/STEPS)
//! * [SimBiology](https://fr.mathworks.com/help/simbio/)
//! * [Copasi](http://copasi.org/)
//! * [BioNetGen](http://bionetgen.org/)
//! * [VCell](http://vcell.org/)
//! * [Smoldyn](http://www.smoldyn.org/)
//! * [KaSim](https://kappalanguage.org/)
//! * [StochPy](https://github.com/SystemsBioinformatics/stochpy)
//! * [BioSimulator.jl](https://github.com/alanderos91/BioSimulator.jl)
//! * [DiffEqJump.jl](https://github.com/SciML/DiffEqJump.jl)
//! * [Gillespie.jl](https://github.com/sdwfrost/Gillespie.jl)
//! * [GillespieSSA2](https://github.com/rcannood/GillespieSSA2)
//! * [Cayenne](https://github.com/quantumbrake/cayenne)
//!
//! ## Seem unmaintained
//!
//! * [Dizzy](http://magnet.systemsbiology.net/software/Dizzy/)
//! * [Cellware](http://www.bii.a-star.edu.sg/achievements/applications/cellware/)
//! * [STOCKS](https://doi.org/10.1093/bioinformatics/18.3.470)
//! * [StochSim](http://lenoverelab.org/perso/lenov/stochsim.html)
//! * [Systems biology toolbox](http://www.sbtoolbox.org/)
//! * [StochKit](https://github.com/StochSS/StochKit) (successor: GillesPy2)
//! * [SmartCell](http://software.crg.es/smartcell/)
//! * [NFsim](http://michaelsneddon.net/nfsim/)
pub use rand;
pub use rand_distr;