# cealign
[](https://github.com/rvhonorato/cealign/actions/workflows/ci.yml)
[](https://app.codacy.com/gh/rvhonorato/cealign/dashboard?utm_source=gh&utm_medium=referral&utm_content=&utm_campaign=Badge_grade)
[](https://crates.io/crates/cealign)
[](https://docs.rs/cealign)
[](https://doi.org/10.5281/zenodo.18892597)
A Rust implementation of the [Combinatorial Extension (CE) algorithm](https://doi.org/10.1093/protein/11.9.739) for protein structure alignment, available as both a command-line tool and a library.
## Usage
### CLI
```
Usage: cealign [OPTIONS] -m <MOBILE> -t <TARGET>
Options:
-m <MOBILE> Path to the mobile structure
-t <TARGET> Path to the target structure
-o, --output Save aligned structures as PDB files (`<name>_aln.pdb`)
-v, --verbose Increase output verbosity
-p, --plot Save the alignment path plot as plot.png [requires --features plot]
-h, --help Print help
-V, --version Print version
```
Align two structures and print the RMSD:
```bash
cealign -m mobile.pdb -t target.pdb
```
Save the aligned structures to disk:
```bash
cealign -m mobile.pdb -t target.pdb -o
```
Save the alignment path plot (requires `--features plot`):
```bash
cealign -m mobile.pdb -t target.pdb -p
```
### Library
```rust
use cealign::ce;
let (mobile, _) = pdbtbx::open("mobile.pdb").unwrap();
let (target, _) = pdbtbx::open("target.pdb").unwrap();
let (aligned_mobile, reference, rmsd, n_aligned) = ce::align(mobile, target, false);
println!("{:.3}", rmsd);
```
## Installation
### CLI
```bash
cargo install cealign
```
To include the optional alignment path plot feature:
```bash
cargo install cealign --features plot
```
> The `plot` feature requires `libfontconfig` on Linux (`sudo apt-get install libfontconfig-dev`).
### Library
Add to your `Cargo.toml`:
```toml
[dependencies]
cealign = "0.1"
```
## Example
Align crambin X-ray structure ([1CRN](https://www.rcsb.org/structure/1CRN)) against the first NMR model of ([1CCM](https://www.rcsb.org/structure/1CCM)).
1CCM is a multi-model NMR ensemble, so we extract model 1 first:
```bash
# Download structures
curl -s https://files.rcsb.org/download/1CRN.pdb -o 1crn.pdb
curl -s https://files.rcsb.org/download/1CCM.pdb -o 1ccm_full.pdb
# Extract model 1 from the NMR ensemble
awk '/^MODEL/ && $2==1{p=1} p; /^ENDMDL/ && p{p=0}' 1ccm_full.pdb > 1ccm_1.pdb
# Align
cealign -m 1crn.pdb -t 1ccm_1.pdb
```
Expected output (RMSD in Å over the aligned Cα atoms):
```
1.331
```
Full run with verbose logging, alignment output, and plot (requires `--features plot`):
```bash
cealign -m 1crn.pdb -t 1ccm_1.pdb -v -o -p
```
```
[DEBUG cealign] Verbose mode enabled
[INFO cealign::ce] Initial RMSD (full): 22.172
[DEBUG cealign::ce] find_path: 20 candidate paths in buffer
[DEBUG cealign::ce] Candidate path: 5 AFPs (40 residues), aligned RMSD = 1.331
...
[INFO cealign::ce] Final RMSD (aligned CA, 40 residues): 1.331
[INFO cealign::ce] Final RMSD (full): 1.921
1.331
Saved: plot.png
Saved: 1crn_aln.pdb and 1ccm_1_aln.pdb
```
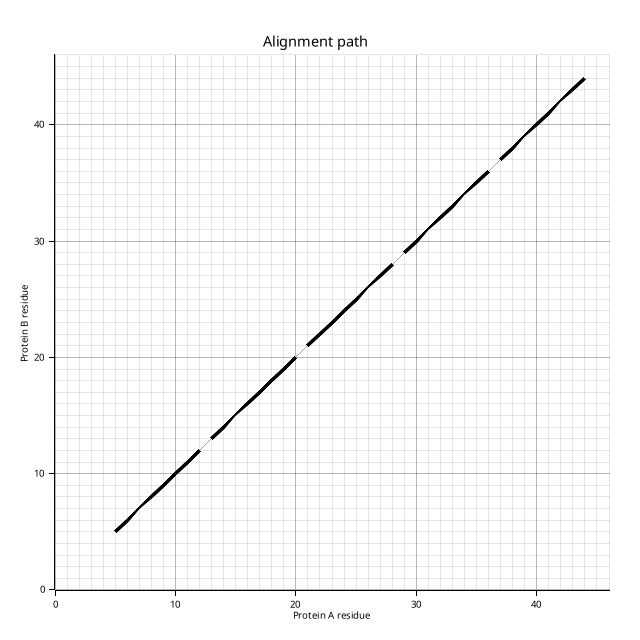
The alignment path plot (`plot.png`) shows each AFP as a diagonal segment on a residue-index grid, mirroring Figure 2 from [Shindyalov & Bourne (1998)](https://doi.org/10.1093/protein/11.9.739):
## Limitations
- Single-model PDB files only — multi-model NMR ensembles must be split before use
- Alignment is performed on Cα atoms; all heavy atoms are transformed using the resulting rotation and translation
## See Also
- [PyMOL cealign](https://pymolwiki.org/index.php/Cealign)
- [Biopython Bio.PDB.cealign](https://biopython.org/docs/dev/api/Bio.PDB.cealign.html)
- [Shindyalov & Bourne (1998)](https://doi.org/10.1093/protein/11.9.739) — original CE algorithm paper
## License
[0BSD](LICENSE)